Description

Nextflow is a reactive workflow framework and a programming DSL
that eases writing computational pipelines with complex data
Software Category: tools
For detailed information, visit the Nextflow website.
Available Versions
The current installation of Nextflow incorporates the most popular packages. To find the available versions and learn how to load them, run:
module spider nextflow
The output of the command shows the available Nextflow module versions.
For detailed information about a particular Nextflow module, including how to load the module, run the module spider command with the module’s full version label. For example:
module spider nextflow/25.04.6
| Module | Version |
Module Load Command |
|---|
| nextflow | 25.04.6 |
module load nextflow/25.04.6
|
Nextflow workflow:
- Nextflow is a workflow management system used to create reproducible and scalable data analyses
- Workflows are written in Groovy and can be deployed in parallel on the HPC system
- Workflows can be executed with modules or containerized environments: Conda or Apptainer
Nextflow processes:
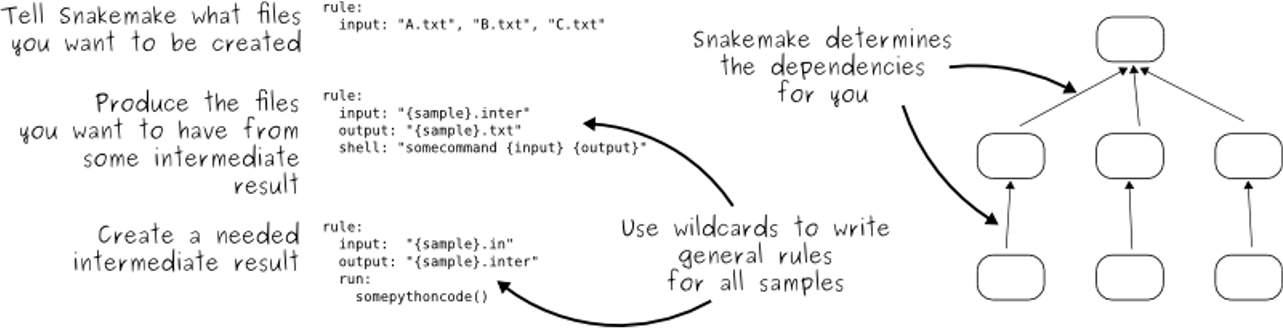
- Snakemake follows the GNU Make paradigm
- Workflows are defined in processes
- Dependencies between the rules are determined automatically, creating a DAG (directed acyclic graph) of jobs that can be parallelized
nextflow.config file:
Config files are generally for
- params: workflow parameters (like input filenames, paths, job settings) processes to define global or process-specific options, or profiles.
- process: additional processes specifying global and/or per-process settings, software environments, and job settings
- profile:
params {
reads = 'sample1.fastq'
adapter = 'AACCGGTT'
ref = 'GCF_000005845.2_ASM584v2_genomic.fna'
outdir = 'results'
}
process {
executor = 'slurm'
queue = 'standard'
clusterOptions = '--account=my-hpc-allocation'
withName: CUTADAPT {
cpus = 2
time = '4h'
mem = '8 GB'
beforeScript = '''
module purge
module load cutadapt
'''
}
withName: BWA_ALIGN {
cpus = 2
time = '4h'
mem = '8 GB'
beforeScript = '''
module purge
module load bwa
module load samtools
'''
}
withName: FREEBAYES {
cpus = 2
time = '4h'
mem = '8 GB'
beforeScript = '''
module purge
module load freebayes
'''
}
}
main.nf:
- The main.nf contains the processes of your workflow (the steps)
- Your workflow will determine the order of the processes in order to create that output
- Each process generally has at least a script, input, output consists of 3 required parts: the input files, the output files, and the shell (command)
- Below is an example of a process to align sequences using hisat. The log and threads options are optional, but included for reference
- The target output is a gene count matrix in a csv format
process CUTADAPT {
publishDir params.outdir, mode: 'copy'
input:
path reads
output:
path "${reads.simpleName}_trimmed.fastq"
script:
"""
cutadapt -a ${params.adapter} -o ${reads.simpleName}_trimmed.fastq $reads
"""
}
process BWA_ALIGN {
publishDir params.outdir, mode: 'copy'
input:
path reads
path ref
output:
path "${reads.simpleName}.bam"
script:
"""
bwa index $ref
bwa mem $ref $reads | samtools sort -o ${reads.simpleName}.bam
"""
}
process FREEBAYES {
publishDir params.outdir, mode: 'copy'
input:
path bam
path ref
output:
path "${bam.simpleName}.vcf"
script:
"""
freebayes -f $ref $bam > ${bam.simpleName}.vcf
"""
}
workflow {
reads_ch = Channel.fromPath(params.reads, checkIfExists: true)
ref_ch = Channel.fromPath(params.ref, checkIfExists: true)
trimmed_ch = CUTADAPT(reads_ch)
bam_ch = BWA_ALIGN(trimmed_ch, ref_ch)
FREEBAYES(bam_ch, ref_ch)
}
- After the rule
align_hisat is completed, the workflow can move to the next rule stringtie_assemble
- Notice that the output of
align_hisat is a .bam file, this is now the input to the rule stringtie_assemble
#```
#rule stringtie_assemble:
genome_gtf=config[‘GENOME_GTF’],
bam=“align_hisat2/{sample}.bam”
output: “stringtie/assembled/{sample}.gtf”
threads: config[‘THREADS’]
shell:
#```
- You can add as many processes as you like as long as they are sequential with inputs and outputs
Slurm for Nextflow:
- The Nextflow pipeline can be executed using a
SLURM script on the HPC system
- Below is an example script to submit to the standard partition with 8 threads
- This script is using a
conda environment called rnaseq
#!/bin/bash
#SBATCH --time=05:00:00
#SBATCH --partition=standard
#SBATCH --mem=4GB
#SBATCH --account=allocation_name
#SBATCH --cpus-per-task=1
module purge
module load nextflow
nextflow run main.nf
Dry Runs:
- Dry-runs are a great way to check your commands before running them
- The code is printed, but not actually run
- For a dry run, use
nextflow run main.nf -dry-run
|
HPC, software, bio
multi-core